Introduction
Il s'agit ici de trouver le minimum d'une fonction. Dans le cas de la modélisation moléculaire, la fonction à minimiser correspond à l'hamiltonien, c'est à dire, la fonction qui permet de retrouver l'énergie de la molécule en fonction de sa géométrie. Pour une molécule de N atomes, la fonction est représentée dans 3N-6 dimensions (N pour l'axe des 'x', N pour l'axe de 'y' et N pour l'axe des 'z', soit 3N, moins 6 dimensions qui correspondent à la rotation (3 dimensions) et à la translation (3 dimensions) de cette molécule). On a donc à faire à une fonction assez complexe pour une molécule organique et extrêmement complexe pour un système biologique de plusieurs milliers d'atomes.
Le principe est de déplacer les atomes afin d'obtenir la conformation la plus stable possible (qui possède une énergie plus basse que le point de départ). Comme son nom l'indique, la minimisation va déplacer les atomes afin de diminuer au fur et à mesure l'énergie de la molécule jusqu'à obtenir une énergie minimale, on finira alors par se trouver dans un puits d'énergie. La difficulté est de trouver la bonne direction à emprunter sur la surface d'énergie, afin de produire la géométrie ayant l'énergie la plus basse possible. Cette partie est donc plus particulièrement basée sur des approches mathématiques, décrivant la façon d'évoluer sur la surface associée à une fonction.
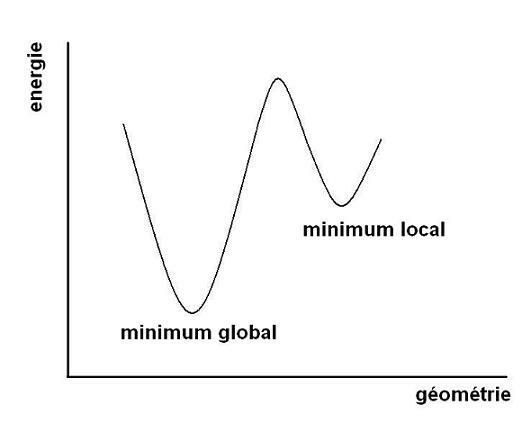
Le minimum global est le point de la surface d'énergie potentielle la plus basse possible, c'est-à-dire le puits le plus profond sur la surface.
Ce point est généralement unique et correspond en principe à la géométrie la plus rencontrée statistiquement par le système moléculaire. Toutefois, Notre surface peut avoir plusieurs minimums locaux qui correspondent à des conformations stables de la molécule, bien qu'elles ne soient pas les plus stables.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.