Exercice de dynamique moléculaire
Partie
Question
Dans cette partie, nous illustrons rapidement le concept de dynamique moléculaire.
Il s'agit de définir un système moléculaire et de simuler ses mouvements à température ambiante (298,15 K). L'objectif, au delà de la visualisation des mouvements moléculaires et inter-moléculaire est d'avoir accès à des grandeurs statistiques. Nous serons alors amenés à visualiser l'évolution de quelques grandeurs sur un tableur (de type excel ou openoffice).
La dynamique moléculaire est illustrée sur un logiciel disponible gratuitement et qui s'installe aisément sur une interface MS Windows. ABALONE est disponible à l'URL suivante : http://www.biomolecular-modeling.com/Abalone/
1/ Nous allons travailler sur un système déjà définit dans le logiciel, ici un petit cluster d'acide cyanhydrique (HCN). Dans « File/Open », choisir le fichier MeCN-5.mlm.
2/ On visualise alors le cluster de 5 molécules de HCN.
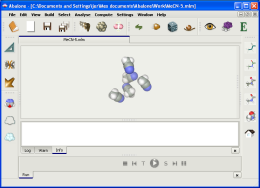
3/ Avant d'envisager une simulation de dynamique moléculaire, il est primordial de s'assurer que le système envisagé correspond à une structure d'énergie minimum. Si ca n'est pas le cas le système sera très instable et les équations du mouvement risquent de diverger en raison de forces de répulsion trop fortes.
4/ lancer une optimisation de géométrie du système. On pourra choisir un algorithme de type « steepest descent », avec un champ de force AMBER03 et des paramètres par défaut.
5/ avec ces paramètres par défaut, le gradient n'est pas atteint, mais le système est suffisamment relaxé pour envisager une simulation de dynamique moléculaire.
6/ Dans l'onglet « Compute », on choisit « Dynamics ». On peut régler la température, qu'on garde à 298,15 K pour l'instant. En revanche, on va réduite le pas d'intégration à 2 fs, ce qui est plus proche de la fréquence de Nyqvist pour un système moléculaire à température ambiante.
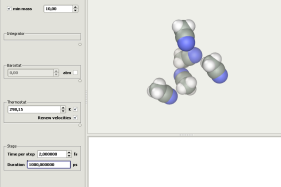
7/ Avant de lancer la simulation, nous allons préciser les grandeurs pour lesquelles nous souhaitons observer l'évolution. Dans l'onglet « Analyse », choisissez « Monitor ».
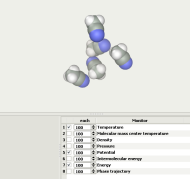
Nous allons réaliser une simulation sans solvant (en phase gazeuse), dès lors, il est inutile de regarder l'évolution de la densité. La pression ne sera pas non plus contrôlée. On s'intéressera uniquement à l'évolution de la température, de l'énergie potentielle et de l'énergie totale.
8/ Lancer le calcul et observez le mouvement des molécules. On observe la rotation des groupements méthyle et des interactions privilégiées entre les atomes chargés positivement (les atomes d'hydrogène) et les atomes chargés négativement (les atomes d'azote). Malgré tout, les molécules ont tendance à s'éloigner en raison d'interactions trop faibles en phase gazeuse.
9/ L'évolution des grandeurs cochées plus haut se trouvent dans des fichiers excel, dans le répertoire « Abalone\Report »
Solution détaillée
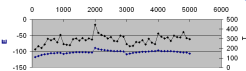
En ouvrant ces fichiers et en traçant l'évolution de chacune de ces grandeurs (attention de remplacer les points par des virgules), on observe que, bien que la température soit théoriquement constante, elle évolue. C'est aussi le cas pour l'énergie totale, comme l'indique cette image. On y lit la température (en noir) sur l'axe de droite et l'énergie totale (en bleu) sur l'axe de gauche. L'objectif est néanmoins atteint puisque la température moyenne du système est de l'ordre de 298 K.
Maintenant, vous pouvez essayer de modifier divers paramètres, afin d'en visualiser l'effet sur la trajectoire du système. Vous pouvez aussi réaliser des simulations sur d'autres systèmes (le C60 par exemple) et analyser l'évolution de distances ou d'angle au fur et à mesure du temps.
Notez que le logiciel propose deux autres tutoriels qui vous permettrons de vous familiariser davantage avec les simulations de dynamique moléculaire.