exercice minimisation
Partie
Question
La minimisation de l'énergie est illustrée sur un logiciel disponible gratuitement et qui s'installe aisément sur une interface MS Windows. ABALONE est disponible à l'URL suivante : http://www.biomolecular-modeling.com/Abalone/
1/ Téléchargez, installez et lancez Abalone.
Vous obtenez une interface graphique qui ressemble à ceci :
2/ Nous allons utiliser une structure déjà existante dans le logiciel. Il s'agit d'une molécule de fullerène, nommée C60. Dans file/open, sélectionner le fichier C60.mlm.
3/ Cliquez sur le bouton pour visualiser la structure en mode « batons » ![]()
Vous obtenez la vue suivante :
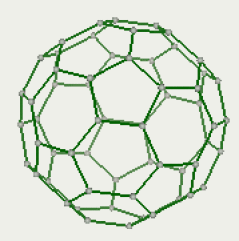
4/ Il faut maintenant assigner un type de champ de force pour pouvoir estimer l'énergie de la molécule. Dans l'onglet « «Settings », cliquez sur « Assign FF types », sélectionnez AMBER03 et validez. Il s'agit d'un champ de force typique de mécanique moléculaire, plus généralement utilisé pour les systèmes biomoléculaires.
5/ A présent, il est possible de calculer l'énergie de cette molécule. Pour cela cliquez sur le bouton ![]()
Dans l'onglet « Info » vous voyez apparaître l'énergie totale ainsi que l'ensemble des termes qui la compose. L'énergie totale vaut 710,863 kcal/mol et on peut par exemple noter que la contribution des termes de liaison (bond) vaut 2,149 kcal/mol.
6/ Dans l'onglet « Analyse », cliquez sur « Gradient ». Le gradient d'un structure stable doit être nul, ou très faible. Ici, le gradient vaut 0,034, indiquant que cette structure ne correspond pas à un minimum d'énergie pour ce champ de force
7/ Assignez un autre champ de force et recalculez ces deux grandeurs. Par exemple, avec le champ de force OPLS, l'énergie vaut 1007,26 kcal/mol et le gradient 0,08. Ceci illustre bien que cette énergie n'a pas de réelle signification. C'est surtout sa variation qui est importante.
8/ Dans l'onglet « Compute », choisissez « Optimise ». Un nouvel onglet s'ouvre, dans lequel il est possile de choisir parmi différents type de «minimiseurs», chacun étant lié à une approche différente. On reconnaît notamment « Conjugate Gradient » (appelé Conjugate Descent) et « Steepest Descent ». Choisir « Steepest Descent », qui est très couramment utilisé en raison de sa rapidité.
9/ Dans l'onglet « Analyse », on choisit « Geometry ». En cliquant sur 2 atomes de carbone directement liés on note que la distance est de 1,4047 Å. On peut modifier cette distance en changeant les coordonnées cartésiennes (x y ou z) d'un des atomes.
10/ En utilisant les paramètres par défaut (Gradient = 0,001, Iterations = 100), avec le champ de force OPLS, lancer l'optimisation en cliquant sur le bouton![]() .
.
On voit la structure évoluer jusqu'à atteindre une géométrie très symétrique, associée à l'énergie minimale (1006,93 kcal/mol).
11/ Quittez puis relisez de nouveau la structure initiale du C60. Lancez une optimisation avec l'approche « Conjugate Descent » avec les mêmes paramètres. On voit que l'optimisation se fait beaucoup plus rapidement (en 10 fois moins d'itérations !), car la structure était très proche de sa structure optimale. Ici, le fait que le calcul est plus long ne se sent pas car les 2 calculs sont très rapides.
Solution détaillée
L'optimisation de la géométrie va dépendre du champ de force ainsi que de la technique utilisés. Néanmoins, quelque soit le type de molécule étudiée, il est absolument primordial de réaliser une optimisation de géométrie avant d'envisager la moindre analyse structurale ou énergétique.