La Méthode Hatree-Fock
Nous avions défini un Hamiltonien électronique approché :
Le corrolaire de cette approximation est alors que la fonction d'onde totale peut-être considérée comme le produit des solutions mono-électroniques de cette équation
On écrit :
Malheureusement, cette fonction d'onde ne satisfait pas au principe de Pauli qui stipule que la fonction d'onde décrivant un système multiélectronique doit changer de signe lors de la permutation des coordonnées de deux électrons quelconques.
Pour obéir au principe de Pauli, il faut introduire une fonction qui permettra de définir les propriétés de spin. \(\chi\) i est une spinorbitale, produit d'une orbitale d'espace \(\psi\) i par sa fonction de spin associée \(\eta\) i.
Dans l'approximation orbitale, la fonction d'onde qui satisfait le principe énoncé ci-dessus se met sous la forme d'un déterminant de Slater tel que :
Nous avons défini de nombreuses approximation qui ont mené à l'écriture d'un hamiltonien simplifié, Hel,approché.
Ici, nous garderons le terme qui définit l'interaction entre les électrons et nous appliquerons cet Hamiltonien aux spinorbitales présentées ci-dessus.
avec \(\psi=|\chi1....\chi n|\)une fonction d'onde multiélectronique à n électrons écrite sous la forme d'un déterminant de Slater construit à partir de n spinorbitales.
Ces équations définissent un opérateur F, appelé Hamiltonien de Hartree-Fock, qui vérifie la relation:
Où l'on définit :
- Ji(1) : opérateur coulombien tel que :
- Ki(1) : opérateur d'échange tel que :
On obtient finalement :
En appliquant le principe variationnel, la meilleure fonction d'onde décrivant le système doit être celle qui correspond à un minimum de l'énergie. L'écriture de cette condition d'extremum de l'énergie par rapport à chaque spinorbitale conduit à un ensemble d'équations appelées équations de Hartree-Fock de la forme:
et qui définissent un ensemble de fonctions parmi lesquelles se trouvent des spinorbitales permettant de construire un déterminant de Slater qui approche le mieux la fonction d'onde multiélectronique du système étudié.
Dans le cas d'un système multiélectronique à couche fermées, c'est-à-dire comportant des orbitales occupées par 2 électrons, les équations de Hartree-Fock peuvent alors se simplifier en équation de Roothan :
Avec :
Ces équation de Roothan peuvent se simplifier sous forme matricielle :
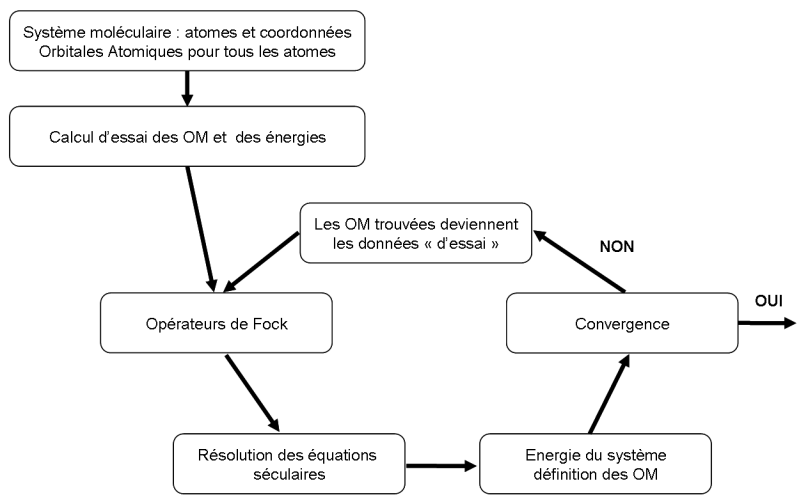
Le déroulement d'un calcul Hartree-Fock peut être résumé selon le schéma suivant :
La limite Hatree-Fock :
Nous avons développé cette méthode Hartree-Fock dans le cadre des approximations initiales, dont la méthode LCAO fait partie. Dans le cadre de cette approximation LCAO, les orbitales moléculaires sont développées comme une combinaison linéaire d'orbitales atomiques. La méthode variationnelle, et donc la méthode Hartree-Fock, dépendent donc directement de la forme que prend cette combinaison linéaire. L'énergie associée, l'énergie Hartree-Fock dépend donc de la façon dont on décrit les orbitales atomiques.
Supposons que l'on obtienne une énergie EA en utilisant une base d'orbitales atomique BA. En utilisant une base d'orbitales atomiques BB, l'énergie EB sera différente de EA, même si le système considéré est le même. De plus, si BB est une base plus étendue, l'énergie EB sera plus petite que EA (plus négative, soit plus grande en valeur absolue).
Ainsi, plus la base utilisée va être étendue (grande), plus l'énergie Hartree-Fock va diminuer pour finalement atteindre une limite que l'on appelle la limite Hartree-Fock.
D'un point de vue pratique, la méthode HF est très utile pour effectuer des premières prédictions de niveau acceptable sur de nombreux systèmes. Elle donne de bons résultats concernant les géométries et le calcul des fréquences d'états stables et de certains états de transition. Par contre, le fait qu'elle ne prenne pas en compte la corrélation électronique la rend impropre dans de nombreux cas : notamment lorsque l'on veut obtenir précisément des énergies de réaction ou de dissociation.