Réactions séries du premier ordre
Approche qualitative
Avant d'aborder dans le détail l'étude analytique de la loi de vitesse, il est utile de se familiariser avec le comportement qualitatif du système cinétique.
\(\mathbf{\textrm A\to\textrm B\to\textrm C}\)
Soient \(k_1\) et \(k_2\) les coefficients de vitesse des deux réactions.
\(C_{\textrm A0}\) la concentration initiale en \(\textrm A\), les concentrations initiales en \(\textrm B\) et \(\textrm C\) étant nulles.
Les réactions étant toutes deux du premier ordre, leurs vitesses s'expriment simplement par :
\(\textrm v_1=k_1.C_\textrm A\) et \(\textrm v_2=k_2.C_\textrm B\)
En ce qui concerne le réactif \(\textrm A\) , il est consommé uniquement par la première réaction.
D'où \(-\frac{\textrm dC_\textrm A}{\textrm dt} = \textrm v_1 = k_1.C_\textrm A\) .
Sa vitesse de transformation n'est pas influencée par la deuxième réaction.
La décroissance de \(\textrm A\) suit donc une loi du premier ordre tout à fait classique.
La vitesse \(\textrm v_1\) de la première réaction diminue donc de manière monotone au cours du temps.
En ce qui concerne le réactif \(\textrm B\) , il est formé par la première réaction et consommé par la seconde.
D'où \(\frac{\textrm dC_\textrm B}{\textrm dt} = \textrm v_1 - \textrm v_2 = k_1.C_\textrm A - k_2.C_\textrm B\) .
La vitesse \(\textrm v_2\) de la deuxième réaction est nulle au temps \(t = 0\) puisque la concentration initiale en \(\textrm B\) est nulle.
Au début de la réaction, cette vitesse augmente au fur et à mesure que la concentration de \(\textrm B\) augmente.
On a donc \(\textrm v_1\) qui diminue tandis que \(\textrm v_2\) augmente, la vitesse de formation de \(\textrm B\), \(\frac{\textrm dC_\textrm B}{\textrm dt}\) diminue donc jusqu'à ce que \(\textrm v_1 = \textrm v_2\).
C'est à dire \(\frac{\textrm dC_\textrm B}{\textrm dt} = \textrm v_1 - \textrm v_2 = 0\).
La concentration de \(\textrm B\) passe donc par un maximum après lequel la vitesse \(\frac{\textrm dC_\textrm B}{\textrm dt}\) devient négative.
Il s'en suit également que la vitesse \(\textrm v_2\) de la deuxième réaction, qui était croissante avant se point, diminue en même temps que \(C_\textrm B\) après son maximum.
En ce qui concerne le réactif \(\textrm C\) , il est formé seulement par la deuxième réaction.
Sa vitesse de formation la deuxième réaction s'écrit donc \(\frac{\textrm dC_\textrm C}{\textrm dt} = \textrm v_2 = k_2.C_\textrm B\) .
Cette vitesse est nulle au temps \(t = 0\), puisque \(C_{\textrm B0} = 0\).
Puis elle augmente avec l'augmentation de \(C_\textrm B\).
Il est à noter que lorsque \(C_\textrm B\) passe par son maximum (donc \(\frac{\textrm dC_\textrm B}{\textrm dt} = 0\)), la courbe \(C_\textrm C = f(t)\) présentera un point d'inflexion, puique \(\frac{\textrm dC_\textrm C}{\textrm dt} = \textrm v_2\) change de signe en ce point.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Voyons qualitativement l'influence des valeurs des coefficients de vitesse \(k_1\) et \(k_2\) sur l'allure des courbes.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Etude analytique de la loi de vitesse
\(\mathbf{\textrm A\to\textrm B\to\textrm C}\)
Soient \(\textrm v_1\) et \(\textrm v_2\) les vitesses et \(k_1\) et \(k_2\) les coefficients de vitesses de la première et de la deuxième réaction.
Au temps \(t = 0\), la concentration initiale de \(\textrm A\) est \(C_{\textrm A0}\) ; les concentrations initiales de \(\textrm B\) et de \(\textrm C\) étant nulle.
Posons \(x\) la concentration de \(\textrm A\) transformée à l'instant \(t\) et \(y\) la concentration de \(\textrm C\) formée au même instant.
Les concentrations courantes à cet instant \(t\) sont respectivement : \(C_\textrm A = C_{\textrm A0} - x\) ; \(C_\textrm B = x - y\) ; \(C_\textrm C = y\) .
On remarque que la stœchiométrie des réactions conduit à : \(C_\textrm A + C_\textrm B + C_\textrm C = C_{\textrm A0}\).
Les vitesses des réactions directe et inverse considérées isolément s'écrivent :
\(\mathbf{\textrm v_1=k_1.C_\textrm A \textrm v_2=k_2.C_\textrm B}\)
Les vitesses de transformation des trois constituants du système sont :
\(\mathbf{\frac{\textrm dC_\textrm A}{\textrm dt} = - \textrm v_1 = - k_1.C_\textrm A}\)
\(\mathbf{\frac{\textrm dC_\textrm B}{\textrm dt} = \textrm v_1-\textrm v_2 = k_1.C_\textrm A-k_2.C_\textrm B}\)
\(\mathbf{\frac{\textrm dC_\textrm C}{\textrm dt} = \textrm v_2 = k_2.C_\textrm B}\)
En introduisant les variables \(x\) et \(y\) introduites ci-dessus, on obtient les lois de vitesses sous leur forme différentielle :
\(\mathbf{\textrm v_1=\frac{\textrm dx}{\textrm dt} = k_1.C_\textrm A= k_1.(C_{\textrm A0}-x)}\)
\(\mathbf{\textrm v_2=\frac{\textrm dy}{\textrm dt} = k_2.C_\textrm B= k_2.(x-y)}\)
On peut intéger indifféremment le premier ou le second système d'équations.
La première équation est celle d'une loi du premier ordre :
\(\mathbf{\frac{\textrm dC_\textrm A}{\textrm dt}=-\textrm v_1=-k_1.C_\textrm A}\) où \(\mathbf{\textrm v_1=\frac{\textrm dx}{\textrm dt}=k_1.(C_{\textrm A0}-x)}\)
et conduit à l'expression de \(C_\textrm A\) en fonction du temps :
\(\mathbf{C_\textrm A=C_{\textrm A0}.\exp(-k_1.t)}\)
En portant cette expression dans la seconde équation, on obtient une équation qui ne comporte plus qu'une variable inconnue (\(C_\textrm B\) ou \(y\)) dont l'intégration donne l'expression de \(C_\textrm B\) en fonction du temps :
\(\mathbf{C_\textrm B=C_{\textrm A0}.\frac{k_1}{k_1-k_2}.\Big(\exp(-k_2.t)-\exp(-k_1.t)\Big)}\)
Enfin, en portant ces expressions dans la relation \(C_\textrm A + C_\textrm B + C_\textrm C = C_{\textrm A0}\) , on tire l'expression de la concentration du dernier constituant \(C_\textrm C\) en fonction du temps :
\(\mathbf{C_\textrm C=C_{\textrm A0}.\Big(1+\frac{k_2.\exp(-k_1.t)-k_1.\exp(-k_2.t)}{k_1-k_2}\Big)}\)
Nota les expressions de \(C_\textrm B\) et \(C_\textrm C\) en fonction du temps, ne sont valable que pour \(k_1\) différent de \(k_2\).
Pour des détails sur l'intégration de la loi de vitesse, cliquez ici[1].
Dans le cas particulier où \(k_1 = k_2\) on obtient des expressions différentes. Mais les conclusions qualitatives développées ici restent valables.
Pour voir les lois de vitesse dans le cas particulier où \(k_1 = k_2\) , cliquez ici[2].
Pour voir uniquement les formules spécifiques dans le cas particulier où \(k_1 = k_2\) , cliquez ici[3].
Remarque :
L'expression de \(C_\textrm A = f(t)\) est simplement une loi du premier ordre. La transformation de \(\textrm A\) selon la première réaction n'est pas influencée par la transformation ultérieure de \(\textrm B\).
Les expressions de \(C_\textrm B\) et \(C_\textrm C\) sont un peu plus complexes. S'il n'est pas nécessaire de les connaître par cœur, il est indispensable de savoir les retrouver dans un formulaire et les utiliser.
Cependant, on peut facilement tirer un certain nombre de conclusions de la forme des lois de vitesses sous leurs diverses formes.
A tout instant, la somme des concentrations est égale à la concentration initiale de \(\textrm A\) : \(\mathbf{C_\textrm A +C_\textrm B + C_\textrm C = C_{\textrm A0}}\) . De même : \(\mathbf{\frac{\textrm dC_\textrm A}{\textrm dt} + \frac{\textrm dC_\textrm B}{\textrm dt} + \frac{\textrm dC_\textrm C}{\textrm dt} = 0}\).
Les courbes \(C_\textrm A\) et \(C_\textrm C\) sont respectivement toujours décroissante et croissante, car \(\textrm A\) est consommé et \(\textrm C\) formé. \(C_\textrm A\) passe de la valeur \(C_{\textrm A0}\) à 0, tandis que \(\textrm C\) passe de 0 à \(C_{\textrm A0}\).
La courbe \(C_\textrm B\) est nulle au temps \(t = 0\) et à la fin de la réaction, elle passe par un maximum au bout d'un temps noté \(t_\textrm{max}\). A cet instant \(\mathbf{\frac{\textrm dC_\textrm B}{\textrm dt} = 0}\) or, la vitesse de formation de \(\textrm C\) s'écrit : \(\mathbf{\frac{\textrm dC_\textrm C}{\textrm dt} = k_2.C_\textrm B}\) ; en différenciant cette équation, il vient : \(\mathbf{\frac{\textrm d^2C_\textrm C}{\textrm dt^2} = k_2.\frac{\textrm dC_\textrm B}{\textrm dt} = 0}\).
Le maximum de \(C_\textrm B\) correspond donc à un point d'inflexion de la courbe \(C_\textrm C\).
L'instant \(t_\textrm{max}\) correspondant au maximum de \(C_\textrm B\) est relié aux coefficients de vitesse par la relation :
\(\mathbf{t_\textrm{max}=\frac{1}{k_1-k_2}.\ln\frac{k_1}{k_2}}\) avec \(k_1\) différent de \(k_2\).
A cet instant, les vitesses des réactions 1 et 2 sont égales. \(\textrm v_1 = \textrm v_2\)
Donc \(k_1.C_\textrm{A,max} = k_2.C_\textrm{B,max}\) ; en appelant \(C_\textrm{A,max}\) la concentration de \(\textrm A\) au temps \(t_\textrm{max}\).
D'où \(\mathbf{\frac{C_\textrm{A,max}}{C_\textrm{B,max}}=\frac{k_2}{k_1}}\) .
Pour obtenir des détails sur l'établissement de ces relations, cliquez ici[4].
Remarque :
Les expressions ci-dessus sont valables pour \(k_1\) différent de \(k_2\) .
Dans le cas particulier où \(k_1 = k_2\) , on trouve : \(\mathbf{t_\textrm{max}=\frac{1}{k_1}}\) et \(\mathbf{C_\textrm{B,max} = C_\textrm{A,max}}\).
Pour obtenir des détails sur l'établissement de ces relations dans le cas particulier où \(k_1 = k_2\) , cliquez ici[5]
Traitement des données expérimentales
\(\mathbf{\textrm A\to\textrm B\to\textrm C}\)
Le coefficient de vitesse \(k_1\) est facile à déterminer car la décroissance de \(\textrm A\) suit une loi du premier ordre. On utilisera donc les méthodes décrites dans le chapitre traitant de l' "Approche expérimentale des lois de vitesse".
Les lois de vitesses intégrées donnant les concentrations \(C_\textrm B\) et \(C_\textrm C\) en fonction du temps ont une forme relativement complexe, difficile à exploiter pour déterminer le coefficient de vitesse \(k_2\). Cependant, en exploitant le point singulier pour lequel \(C_\textrm B\) passe par son maximum, il est facile de déterminer le rapport \(\frac{k_1}{k_2}\).
Au contraire, les lois de vitesse sous leur forme différentielles sont simples et permettent une exploitation immédiate.
Exemple : Exploitation des vitesses
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Exemple : Analyse à partir de CBmax
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Cas particuliers limites
\(\mathbf{\textrm A\to\textrm B\to\textrm C}\)
Lorsque le rapport \(\mathbf{\frac{k_1}{k_2}}\) est beaucoup plus grand que 1, la première réaction peut être considérée comme complètement terminée, alors que l'effet de la seconde ne s'est pas encore fait sentir.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
On peut donc considérer que la réaction s'effectue en deux étapes successives nettement distinctes et étudier chacune d'elle séparément.
Lorsque le rapport \(\mathbf{\frac{k_1}{k_2}}\) est beaucoup plus petit que 1, la seconde réaction est plus rapide que la première. L'intermédiaire \(\textrm B\) peut ne peut pas s'accumuler. Sa concentration passe par son maximum pour des temps faibles (inférieurs au temps de demie-vie de \(\textrm A\)). La formation de \(\textrm C\) est limitée par concentration de \(\textrm B\) qui reste faible. Lorsque \(\textrm A\) a pratiquement disparu, \(\textrm C\) déjà est proche d'atteindre sa valeur limite.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
On peut alors, sous certaines conditions, considérer qu'après une phase d'induction, la concentration de l'intermédiaire \(\textrm B\) reste quasiment constante. Le traitement des résultats expérimentaux s'en trouve alors grandement simplifié.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Lorsque le rapport \(\mathbf{\frac{k_1}{k_2}}\) est égal à 1, les expressions générales des lois de vitesses intégrées concernant \(C_\textrm B\) et \(C_\textrm C\) ne sont plus applicables (car elles font intervenir à leur dénominateur la différence \(k_1 - k_2\)). L'expression de \(C_\textrm A = f(t)\) reste évidemment applicable puisqu'elle ne fait intervenir que \(k_1\) de même que le rapport \(\frac{C_\textrm{A,max}}{C_\textrm{B,max}} = \frac{k_1}{k_2}\) qui vaut alors 1. Il faut utiliser des formules spécifiques.
Pour voir les formules spécifiques au cas particulier où \(k_1 = k_2\), cliquez ici[2].
Exemples
L'exemple le plus simple nous est donné par les désintégrations radioactives au cours desquelles, un radioélément \(\textrm E_1\) se désintègre pour donner un autre radioélément \(\textrm E_2\), lequel se désintègre à son tour, constituant ainsi une famille radioactive :
\(\mathbf{\textrm E_1\stackrel{k_1}{\to}\textrm E_2\stackrel{k_2}{\to}\textrm E_3\stackrel{k_3}{\to}\textrm E_4\stackrel{k_4}{\to}\textrm E_5 ...}\)
Exemple : Une partie de la famille radioactive issue de l'uranium 238
\(\mathbf{\textrm{}^{238}\textrm U\to\textrm{}^{234}\textrm{Th}\to\textrm{}^{234}\textrm{Pa}\to\textrm{}^{234}\textrm U\to\textrm{}^{230}\textrm{Th}\to\textrm{}^{226}\textrm{Ra}\to\textrm{}^{222}\textrm{Rn}}\)
Il est à noter que les coefficients de vitesses, appelés dans ce cas constantes de désintégration; peuvent avoir des valeurs qui varient de plusieurs ordres de grandeurs. Par exemple la période (ou temps de demie vie) de l'uranium 238 est de 4,5.109 ans, tandis celle de l'iode 131 est de 8 jours et du polonium 214 de 1,6.10-4 secondes. Rappelons que les coefficients de vitesse sont reliés aux périodes \(t_{\frac12}\) par la relation :
\(k=\frac{\ln2}{t_{\frac12}}\)
D'où les valeurs des coefficients de vitesse pour ces nucleides :
\(\textrm{}^{238}\textrm U k=\textrm{4,9}.10^{-18} \textrm s^{-1}\)
\(\textrm{}^{131}\textrm I k=\textrm{1,0}.10^{-6} \textrm s^{-1}\)
\(\textrm{}^{214}\textrm{Po} k=\textrm{4,3}.10^{+3} \textrm s^{-1}\)
Il y a donc un rapport de 1021 entre la valeur du coefficient de vitesse de \(\textrm{}^{238}\textrm U\) et celle de \(\textrm{}^{214}\textrm{Po}\) .
Exemple : Les composés polyfonctionnels
Les composés polyfonctionnels peuvent aussi subir des transformations successives affectant chacune de leurs fonctions.
Ainsi, l'hydrolyse d'un diester symétrique, se fait en deux étapes: hydrolyse de la première fonction ester suivie de l'hydrolyse de la seconde.
En toute rigueur, nous sommes ici en présence d'une réaction complexe puisqu'elle est constituée de deux réactions mais qu'il y a un réactif commun (\(\textrm H_2\textrm O\) ou \(\textrm{HO}^-\) si on est en milieu basique) qui intervient dans les deux étapes.
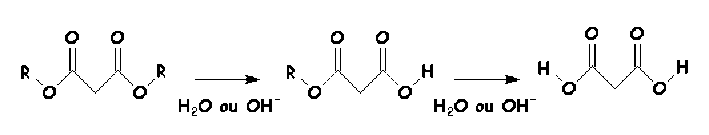
La réaction est bien série par rapport au monoester (qui est le produit de la première réaction et le réactif de la seconde), elle est aussi parallèle par rapport à l'eau (qui est un réactif pour les deux réactions). Cependant, si la quantité d'eau (ou d'ion hydroxyle si on est en milieu basique) est en quantité largement supérieure à celle du diester on peut considérer que les réactions sont dégénérée. Dans ces conditions les deux réactions obéissent à une loi d'ordre 1 apparent .
Pour un rappel sur la dégénérescence d'ordre , se reporter à la partie "Concepts fondamentaux - Dégénérescence de l'ordre d'une réaction".