Déplacements chimiques
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Les protons benzéniques apparaissent en général vers 7,3 ppm. Quand le noyau benzénique est substitué, on observe selon la nature du groupement attaché au noyau un glissement du déplacement chimique d soit vers les champs faibles, soit vers les champs forts. Qu'en est-il exactement ?
D'une manière générale, les règles d'Holleman (bien connues en chimie organique) qui régissent la substitution électrophile du noyau aromatique, définissent deux grands types de substituants :
les substituants activants, ortho et para orienteurs,
les substituants désactivants, méta orienteurs.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Les substituants activants, ortho et para orienteurs.
Ce sont les groupements donneurs par conjugaison qui augmentent la densité électronique de l'ensemble des carbones du noyau plus particulièrement en ortho et para. Exemple : \(\textrm{-NH}_2\), \(\textrm{-NHR}\), \(\textrm{-NR}_2\), \(\textrm{-OH}\), \(\textrm{-OR}\)...
Ces groupements sont blindants pour les hydrogènes situés en position ortho et para. Ces protons vont donc apparaître à des champs plus forts et par suite à des déplacements chimiques plus faibles.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Les substituants désactivants, méta orienteurs.
Ce sont les groupements attracteurs par conjugaison qui réduisent la densité électronique de l'ensemble des carbones du noyau plus particulièrement en ortho et para (et par suite la substitution électrophile s'effectue en méta). Exemple : \(\textrm{-(C=O)-H}\), \(\textrm{-(C=O)-OH}\), \(\textrm{-(C=O)-OR}\), \(\textrm{-NO}_2\)...
Ces groupements sont déblindants pour les hydrogènes situés en position ortho et para. Ces protons vont donc apparaître à des champs plus faibles et par suite à des déplacements chimiques plus élevés. Ce déblindage en ortho est souvent très fort car l'effet est directement transmis par la double liaison du groupement.
Notez que pour illustrer ces effets des substituants sur les déplacements chimiques des protons aromatiques, nous avons retenu des dérivés disubstitués en para avec le même substituant. De ce fait, la symétrie de la molécule entraîne l'isochronie des quatre hydrogènes du noyau aromatique. Cela permet d'observer un singulet comme signal ce qui simplifie les comparaisons des déplacements chimiques. Nous verrons plus tard les problèmes posés par les différents couplages observables au niveau du noyau aromatique.
Retenez que :
les groupements donneurs par effet inductif (\(\textrm{CH}_3\textrm-\)) ou par conjugaison augmentent la densité électronique et blindent en ortho, donc déplacement chimique faible, exemple : \(\textrm{-NH}_2\), \(\textrm{-NHR}\),\( \textrm{-NR}_2\), \(\textrm{-OH}\),\( \textrm{-OR}\)...
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
les groupements attracteurs par conjugaison réduisent la densité électronique et déblindent fortement en ortho, donc déplacement chimique fort, exemple : \(\textrm{-(C=O)-H}\),\( \textrm{-(C=O)-OH}\), \(\textrm{-(C=O)-OR}\), \(\textrm{-NO}_2\)...
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Exemple : Autres exemples.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Il est intéressant de remarquer que dans le cas du dérivé ortho du diacétylbenzène, on n'observe pas du tout un fort déblindage du signal des protons aromatiques. Voyons pourquoi...
D'abord il est à noter que l'on observe une pseudoisochronie des protons situés en alpha et en béta des groupements \(\textrm{CO-CH}_3\). On devrait avoir des déplacements chimiques différents... Si on regarde les effets des substituants
\(\delta_{\textrm{Halpha}}\) = effet ortho + effet méta
\(\delta_{\textrm{Hb\'eta}}\) = effet para + effet méta
Il semble que l'effet ortho soit ici voisin de l'effet para. Globalement les protons subissent les mêmes types d'effets des deux substituants... D'où la pseudoisochronie observée.
Par ailleurs, au niveau des déplacements chimiques des protons aromatiques, on peut s'étonner de la faible valeur observée... En fait, l'examen du modèle moléculaire montre qu'il n'est pas plan à cause de la gêne stérique des deux méthyles et de ce fait la transmission des effets de conjugaison ne se fait pas. D'où ce déplacement chimique "anormalement" peu déblindé pour ces protons aromatiques.
Effets des halogènes :
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.
En ce qui concerne les halogènes, fluor, chlore, brome et iode, ils ont de faibles contributions qui ne permettent pas de les classer dans les familles précédentes.
Le fluor a un effet blindant (surtout un couplage observable que l'on étudiera plus tard...), le chlore a un effet quasiment nul ! le brome peu d'effet également... l'iode a peu d'effet aussi.
Système de calcul des déplacements chimiques à base d'incréments :
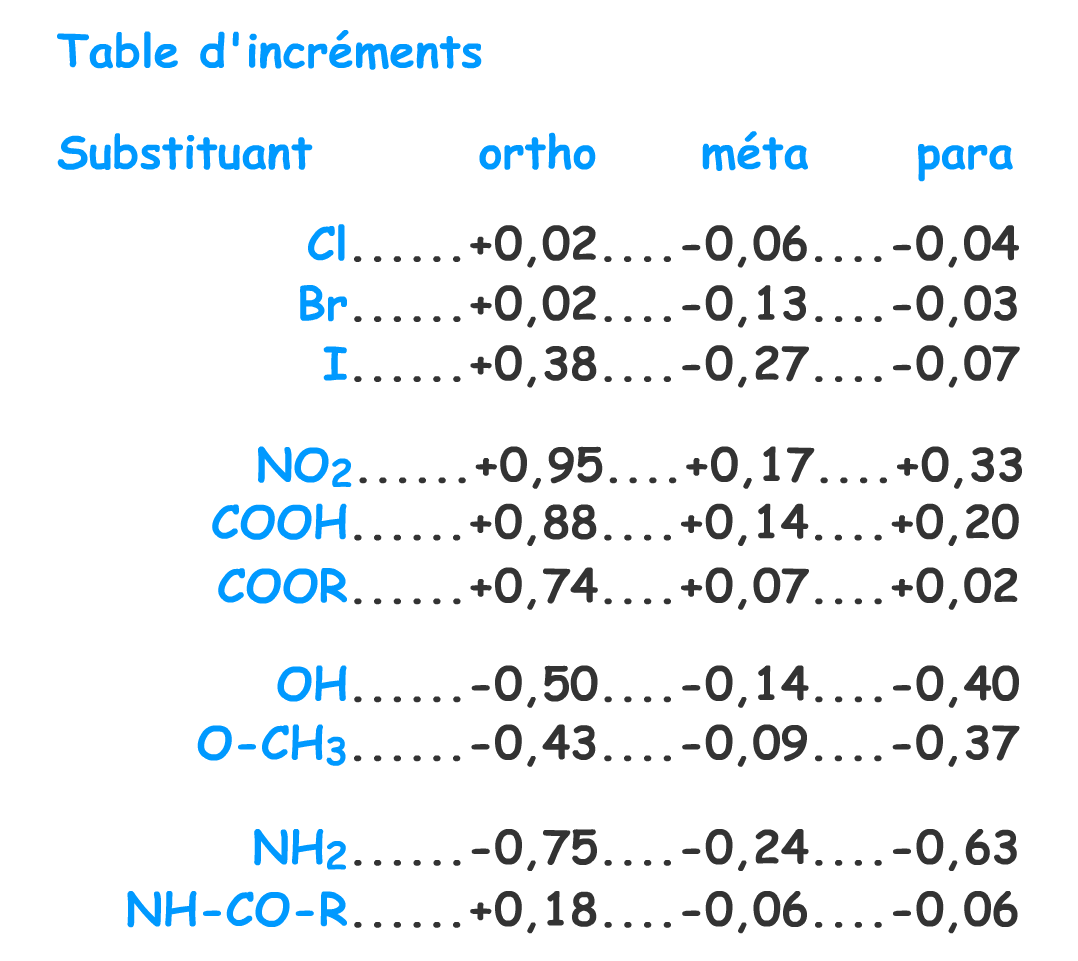
Vous pourrez trouver des tables d'incréments dans la littérature. Ces incréments ont été calculés à partir des spectres des composés monosubstitués.
Selon la position du substituant et selon la nature du substituant, ces incréments sont à additionner ou à retrancher à la valeur 7,27 ppm valeur habituellement observée pour un proton aromatique.
Ces tables permettent de faire un calcul du déplacement chimique d'un proton aromatique en fonction des substituants portés par le noyau et de leurs positions relatives par rapport à ce proton. Il s'agit bien entendu d'une estimation du déplacement chimique et le résultat indique une tendance par rapport à la valeur observée expérimentalement.
Exemple : Exemple de calcul
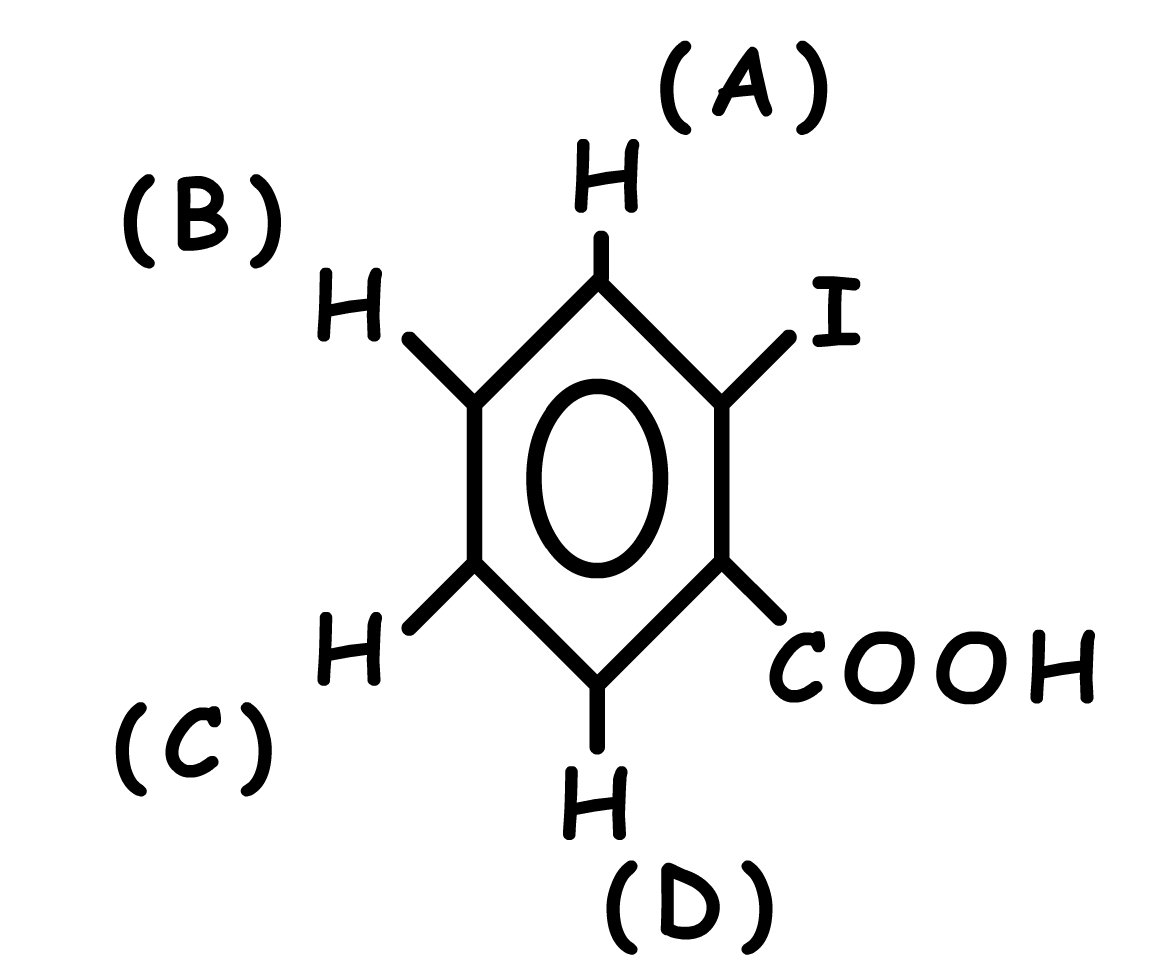
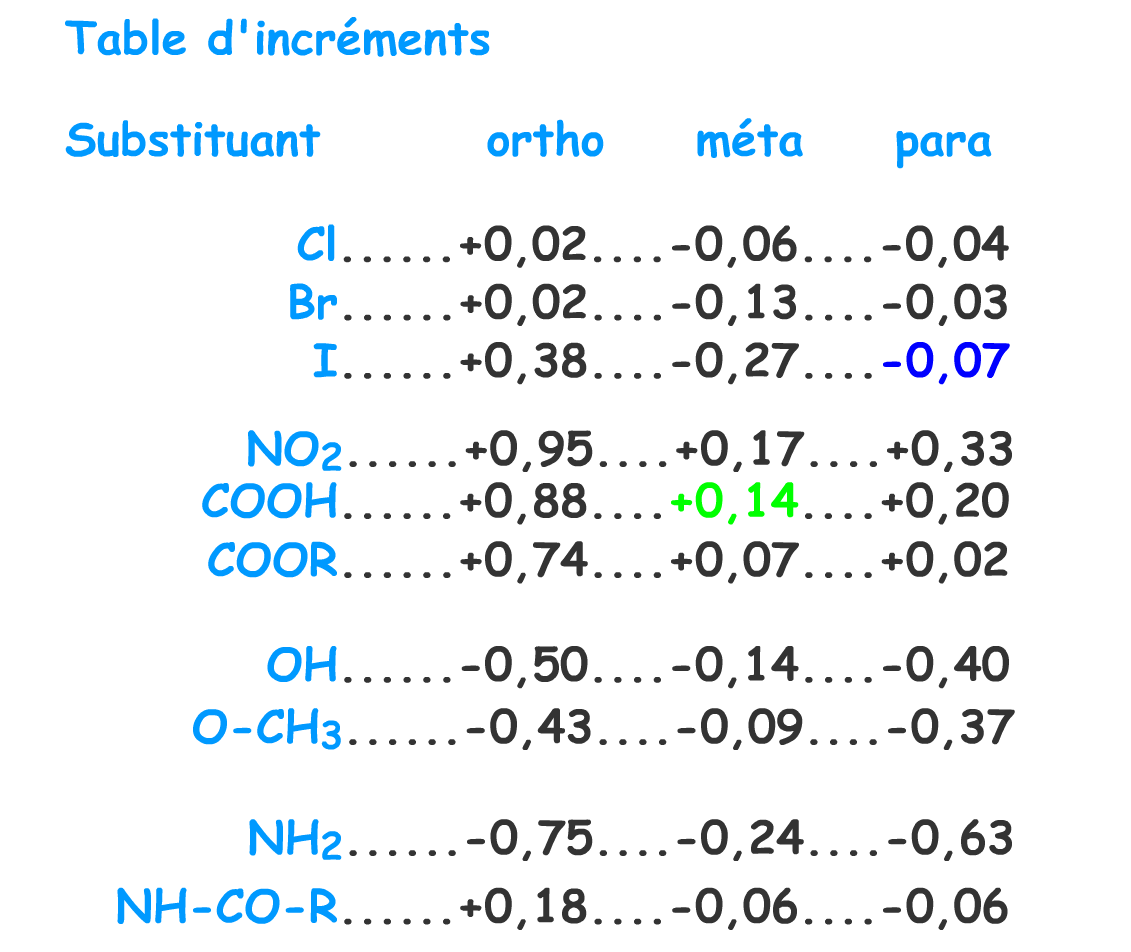
Estimation du déplacement chimique du proton \(\textrm C\) :
\(\textrm H\) aromatique 7,27 ppm
\(\textrm H\) en position para de l'iode - 0,07
\(\textrm H\) en position méta du \(\textrm{COOH}\) + 0,14
Total, d estimé pour \(\textrm C\) : \(\delta\) = 7,27 - 0,07 + 0,14 = 7,34 ppm
Valeur expérimentale observée 7,32 ppm
Vérifiez les valeurs des autres protons aromatiques...
\(\delta\) estimé pour A : 7,90 ppm
\(\delta\) estimé pour B : 7,23 ppm
\(\delta\) estimé pour D : 8,06 ppm
C'est sur ce principe de calcul qu'est basé le simulateur de calcul des déplacements chimiques des protons aromatiques qui est dans la tables des déplacements chimiques.
Entraînez-vous à utiliser le simulateur de calcul des déplacements chimiques du noyau aromatique.
Impossible d'accéder à la ressource audio ou vidéo à l'adresse :
La ressource n'est plus disponible ou vous n'êtes pas autorisé à y accéder. Veuillez vérifier votre accès puis recharger le média.