Bases structurales
Les propriétés des oses reposent sur trois particularités structurales.
Attention :
Les notions suivantes sont au programme de Chimie organique, et doivent être maîtrisées pour aborder ce chapitre.
Polarité du carbonyle
Le carbonyle (double liaison C=O) est fréquemment considéré comme la fonction la plus importante en chimie organique, ainsi qu'en biochimie. L'oxygène est plus électronégatif que le carbone. La double liaison est donc polarisée, avec une charge positive partielle (δ+ ) sur le carbone, et une charge négative partielle (δ- ) sur l'oxygène.
Acidité de l'hydrogène porté par le carbone α
L'hydrogène porté par un carbone immédiatement adjacent au carbonyle (carbone α) est acide du fait de la capacité de l'oxygène voisin à supporter l'excès de densité électronique de la base conjuguée.
Polarité des alcools
L'électronégativité de l'oxygène est à l'origine d'une distribution asymétrique des charges partielles sur O et H dans les alcools. L'hydroxyle -O-H est un dipôle (O)δ- (H)δ+ .
La réactivité du carbonyle est dominée par les processus d'addition.
Comme toute double liaison, le carbonyle est sensible aux additions. Parmi les réactions connues, on peut citer l'hydrogénation catalytique qui conduit à la formation de polyalcools. Dans les additions ioniques, les réactifs polaires s'ajoutent au carbonyle en accord avec la loi de Coulomb : les nucléophiles se lient au carbone électropositif et les électrophiles à l'oxygène électronégatif. C'est le cas des hydrures (NaBH4 par exemple) qui sont capables de réduire le carbonyle mais pas la fonction alcène. Dans le cas des hydrures, le nucléophile (H-) est une base forte et les réactions forment irréversiblement des polyalcools (alditols). Ces réactions sont expliquées en détail au chapitre Réduction des oses.
Des additions sont également possibles avec des bases plus faibles. Ces additions sont réversibles et mènent à des équilibres qui peuvent être déplacés dans les deux sens, en fonction des conditions réactionnelles. Ces réactions, dont fait partie la cyclisation des oses, sont expliquées en détail au chapitre Additions réversibles.
La tautomérie céto-énolique explique des cas d'isomérisation.
L'acidité de l'hydrogène porté par le carbone α est à l'origine d'une tautomérie céto-énolique, processus par lequel le carbonyle se déplace entre le C1 et le C2 d'un ose. Elle permet d'expliquer l'isomérisation réversible d'un cétose en aldose, ainsi que les réactions d'épimérisation en C2 des aldoses. Ces réactions sont expliquées en détail au chapitre Isomérisation.
Les isomérisations peuvent avoir lieu en solution, en présence d'acides ou de bases faisant office de catalyseurs. Dans la cellule vivante, elles sont le fait de réactions enzymatiques considérablement plus rapides et plus spécifiques.
Attention :
Toutes les réactions annoncées ci-dessus sont spécifiques de fonctions (aldéhyde, cétone) portées par les formes linéaires des oses.
Il est intéressant de noter qu'en solution, pour un grand nombre d'oses, ces fonctions sont extrêmement minoritaires dans la mesure où la cyclisation est possible. Toutefois, le principe de la loi d'action de masse et les équilibres qui en découlent permettent d'expliquer que la majorité des molécules s'impliquent totalement dans ces réactions, même quand les formes chimiques nécessaires sont minoritaires.
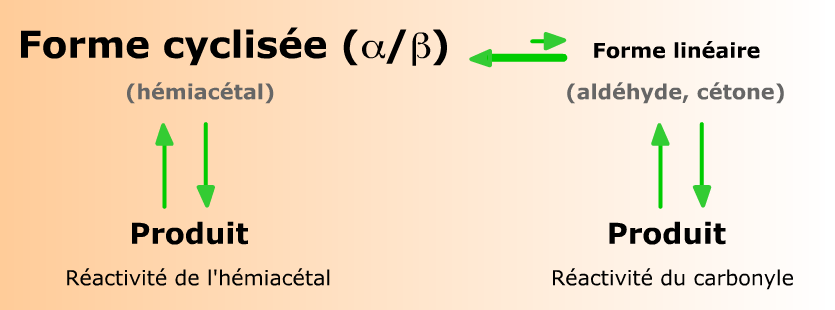
Complément :
La plupart des oses existent sous deux formes, linéaire (minoritaire) et cyclisée (majoritaire). Toute consommation de l'une de ces formes par une réaction entraîne un déplacement de l'équilibre qui régit leurs concentrations respectives. Ainsi, une réaction spécifique de la forme linéaire peut-elle consommer la totalité des molécules d'ose, dans les conditions favorables.
Les alcools autorisent la formation de nombreux éthers et esters, dont certains sont des dérivés naturels d'oses.
Les alcools polaires forment de nombreuses liaisons hydrogène avec l'eau, ce qui fait des oses des molécules organiques hydrophiles et très solubles dans l'eau.
Les oses peuvent aussi former des éthers et des esters. Au laboratoire, cette possibilité est exploitée expérimentalement pour déterminer la structure de sucres complexes, formés d'enchaînements covalents de monosaccharides (oligosaccharides, polysaccharides). Ces réactions sont détaillées au chapitre Ethers et esters, et leur exploitation technique fait l'objet du chapitre Analyse structurale des oligosaccharides.
Il existe des esters naturels, notamment avec les acides phosphorique et sulfurique. Les oses phosphorylés sont décrits parmi les Dérivés naturels d'oses (voir Oses phosphorylés) et les oses sulfatés sont fréquents dans les hétéropolysaccharides (voir Glycosaminoglycanes). Quelques éthers naturels sont également connus dans le règne végétal (voir Ethers et esters).