Additions réversibles
Addition d'alcool
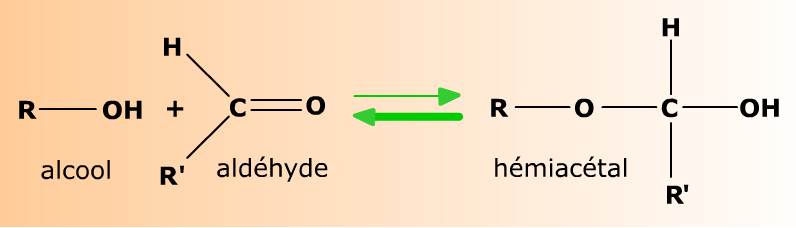
Aldéhydes et alcools réagissent en formant des hémiacétals.
Aldéhydes et alcools forment réversiblement des produits d'addition appelés hémiacétals (parce qu'ils sont à mi-parcours sur la voie réactionnelle menant aux acétals). Les bases structurales de cette addition sont décrites au chapitre Bases structurales.
Complément :
Un alcool R-OH s'ajoute à l'aldéhyde R'-CHO. Le carbone sp2 de l'aldéhyde devient le carbone sp3 de l'hémiacétal, lié à deux atomes d'oxygène. L'équilibre de la réaction est nettement en faveur de l'aldéhyde et de l'alcool.
Complément : Simuler
Simuler le mécanisme chimique de formation d'un hémiacétal
Une animation interactive permet de comprendre le mécanisme réactionnel, étape par étape.
Ce type d'addition est gouverné par un équilibre favorable à l'aldéhyde et à l'alcool, car l'hémiacétal représente une structure instable où un même carbone sp3 est lié à deux hétéroatomes (-OH et R-O-). Un hémiacétal est donc normalement rare et difficile à isoler. La cyclisation des oses ( voir Conditions de la cyclisation) est une exception à cette règle car la formation d'un hémiacétal cyclique apporte un avantage structural significatif à la molécule qui le contient. Toutefois, la facilité relative avec laquelle un hémiacétal se rompt en aldéhyde et alcool signifie qu'un système hémiacétalique est toujours un site de faiblesse structurale dans une molécule, même dans un ose, comme il apparaît au chapitre Mutarotation.
Le système hémiacétalique d'un aldose (en rouge)
La cyclisation d'un aldose implique l'addition d'un alcool R'-OH sur un aldéhyde R-CHO.
R et R' appartenant à la même molécule, il y a formation d'un hémiacétal cyclique.
Le système hémiacétalique d'un cétose (en rouge)
La cyclisation d'un cétose implique l'addition d'un alcool R'-OH sur une cétone R-CO-R". R, R' et R" appartenant à la même molécule, il y a formation d'un hémiacétal cyclique.
Complément : Simuler
Simuler le mécanisme de cyclisation du glucose
Une animation interactive permet de comprendre le mécanisme réactionnel, étape par étape.
Les cétones aussi forment des hémiacétals.
Les cétones se comportent à peu près comme les aldéhydes vis à vis des alcools. Les produits d'addition étaient anciennement appelés hémicétals, mais le terme d'hémiacétal est maintenant admis, quelle que soit la fonction de départ, aldéhyde ou cétone. L'addition d'alcool sur une cétone n'est cependant pas aussi aisée que sur un aldéhyde et l'équilibre est encore moins en faveur de l'hémiacétal. Parmi les sucres toutefois, les cétohexoses montrent, à l'état libre, des systèmes hémiacétaliques internes relativement stables.
La page suivante traite de la formation d'acétals en présence d'un excès d'alcool...
Formation d'acétals
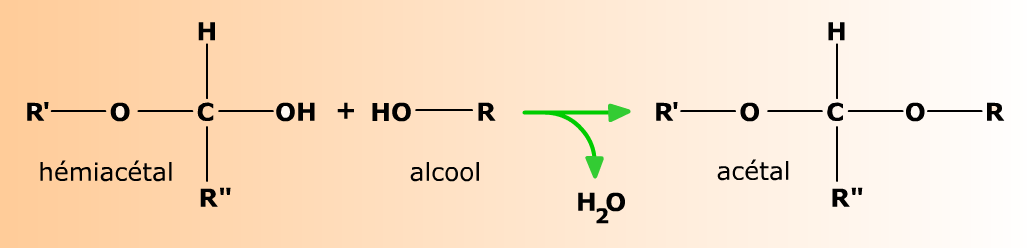
Les hémiacétals se condensent avec les alcools pour former des acétals.
La réaction des aldéhydes et des cétones peut aller au delà de la formation d'hémiacétals, et conduit en milieu légèrement acide à la formation d'acétals. La réaction produit une molécule d'eau; c'est une condensation.
Complément :
En milieu très légèrement acide (HCl 0,25%), un alcool R-OH comme le méthanol se condense à l'hémiacétal pour former un acétal.
Les acétals se forment plus facilement que les éthers, avec lesquels ont les confond souvent. En règle générale, quand deux groupes fonctionnels sont très proches l'un de l'autre dans une même molécule, chacun modifie les propriétés de l'autre. Ici, O-R' rend le groupe -OH voisin plus réactif pour la condensation.
Complément : Simuler
Simuler le mécanisme chimique de formation d'un acétal
Une animation interactive permet de comprendre le mécanisme réactionnel, étape par étape.
Les acétals sont stables en milieu neutre, bien que leur formation soit encore en équilibre réversible. Dans l'eau, les acides (et certaines enzymes) catalysent l'hydrolyse de ces composés.
La formation d'un acétal verrouille la configuration anomérique d'un ose.
Dans le cas des oses, la formation d'un acétal produit un oside, ou O-glycoside, dont la conformation anomérique est celle de l'ose au moment de la réaction. Un O-glycoside n'a pas de pouvoir réducteur (il ne réduit pas les oxydes métalliques) et n'est pas capable de mutarotation. L'acétal étant stable, la configuration anomérique d'un O-glycoside ne change pas au cours du temps.
Complément :
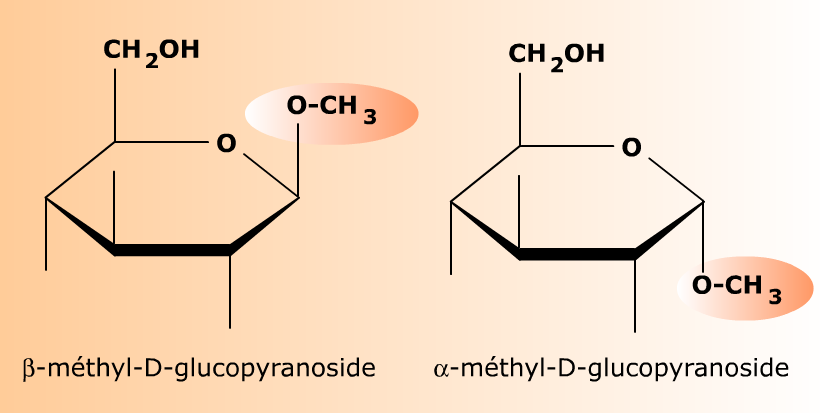
Le D-glucopyranose (hémiacétal) réagit avec le méthanol CH3OH pour former deux O-glycosides (acétals) correspondant aux deux formes anomères α et β du glucose initial. Ces deux osides n'ont pas de pouvoir réducteur et ne sont plus interconvertibles par mutarotation. Le groupement méthyle -CH3 donné par le méthanol constitue l'aglycone des osides.
La partie de la molécule de O-glycoside qui n'appartient pas à l'ose d'origine est appelée aglycone. Les noms des O-glycosides dérivent directement des noms des oses d'origine. Ainsi, le D-glucopyranose donne des D-glucopyranosides, le D-mannopyranose des D-mannopyranosides, le D-ribofuranose des D-ribofuranosides, etc... De nombreux O-glycosides entrent dans la composition des glycolipides.
Les disaccharides sont des O-glycosides dont l'aglycone est un ose.
Portant de nombreuses fonctions alcool, les oses peuvent parfaitement servir d'aglycones à d'autres oses. Les molécules où deux oses sont ainsi liés par une liaison O-glycosidique sont appelées disaccharides, ou encore diholosides (du grec holos = entier) quand les deux oses sont identiques.
Complément :
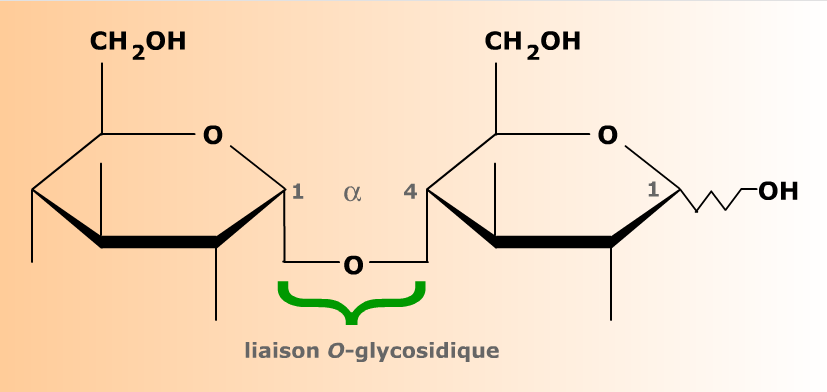
Le maltose est un O-glucopyranoside dont l'aglycone est un résidu de D-glucopyranose. Le résidu d'ose dont le carbone anomérique est pris par la liaison glucosidique (à gauche) n'est plus capable de mutarotation. Sa configuration anomérique est bloquée en α. L'ose dont le carbone anomérique est libre (à droite) est toujours réducteur et capable de mutarotation. Ces propriétés sont symbolisées par la liaison en zig-zag de l'hydroxyle anomérique.
Attention :
Notez bien ! :
L'établissement de liaisons O-glycosidiques entre résidus d'oses (ou de leurs dérivés naturels) est à la base de la structure des glucides complexes, oligo- et polysaccharides (voir Liaisons O-glycosidiques). Toutefois, le mécanisme de formation de ces liaisons in vivoest différent et plus complexe qu'au laboratoire de chimie, même s'il aboutit au même résultat final.
La page suivante traite de l'addition réversible d'amines sur le carbonyle des oses...
Addition d'amines
Les aldéhydes et les cétones se condensent avec les amines primaires pour donner des imines.
Les amines primaires donnent avec les aldéhydes et les cétones le même type de réaction d'addition que les alcools. Le produit formé, appelé carbinolamine (ou hémiaminal) est l'équivalent azoté de l'hémiacétal. Très instables toutefois, les carbinolamines se déshydratent spontanément en donnant des imines, caractérisées par la double liaison C=N. La réaction produisant une molécule d'eau, c'est une condensation.
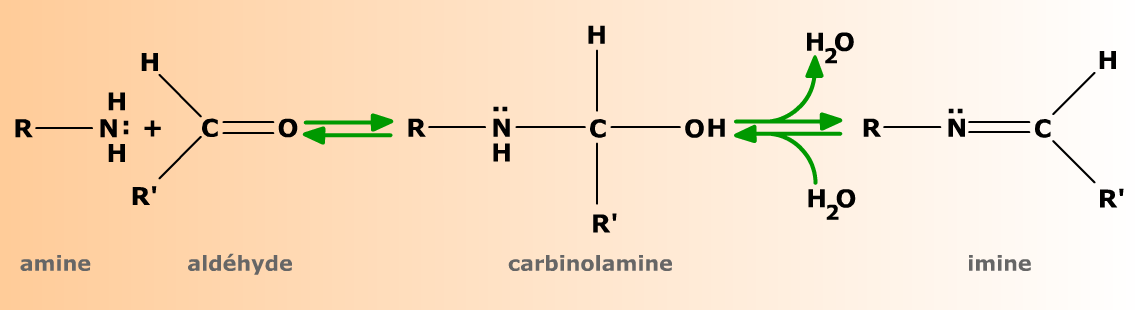
Complément :
L'addition d'une amine primaire R-NH2 sur un aldéhyde R'-CHO donne une carbinolamine (ou hémiaminal) qui se déshydrate spontanément en imine.
La réaction est réversible et peut être orientée dans un sens ou dans l'autre par les conditions expérimentales (pH, température, concentrations de réactifs).
Complément : Simuler
Simuler le mécanisme chimique de formation d'une imine
Une animation interactive permet de comprendre le mécanisme réactionnel, étape par étape.
Les N-glycosides sont des imines cyclisées.
Les imines sont structuralement très semblables au carbonyle et présentent comme lui une polarité due à la différence d'électronégativité des deux atomes doublement liés. Comme les aldoses et les cétoses, les imines formées par les oses peuvent donner lieu à cyclisation, et tendent en solution aqueuse vers un équilibre anomérique (mutarotation), avec des formes anomères α et β.
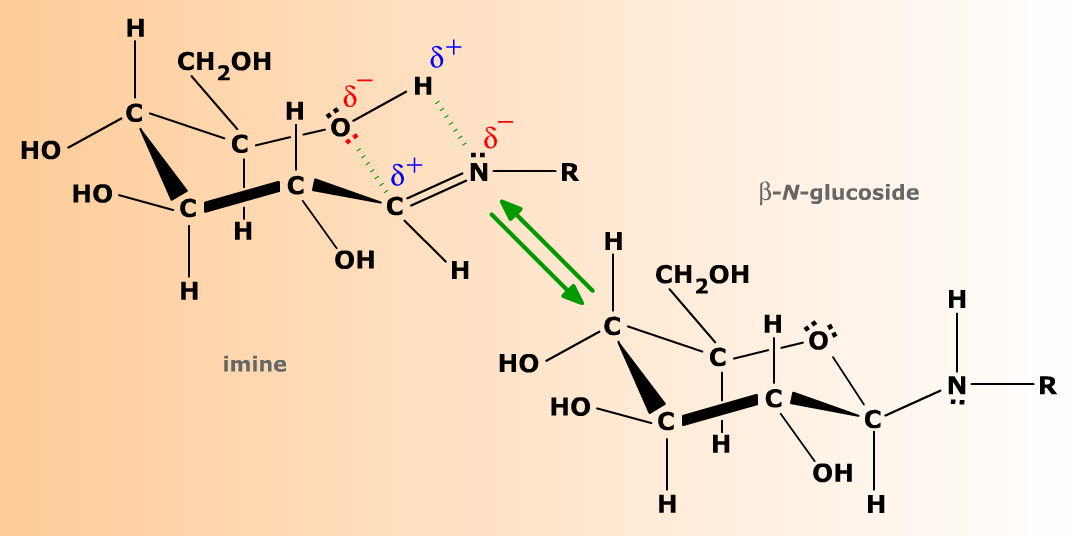
Complément :
La polarité de la double liaison C=N permet aux imines d'oses de se cycliser réversiblement, avec un équilibre anomérique entre formes α et β.
On appelle ces formes cyclisées des glycosylamines N-substituées, ou encore N-glycosides. Comme les O-glycosides, les N-glycosides entrent dans la composition de nombreuses molécules biologiques, dont les plus connues sont les nucléosides et les nucléotides, constitutifs des acides nucléiques.