Prise en main des logiciels
Partie
Durant les 6 TP proposés, qui vont vous permettre de mettre en pratique les notions vues dans le cours, deux logiciels seront utilisés :
- GAMESS, logiciel gratuit, qui permet d'effectuer des calculs de chimie quantique.
- Gabedit, logiciel de visualisation et d'édition de molécules, qui permettra également la préparation des fichiers d'entrée (input) pour GAMESS.
Ces deux logiciels se trouvent facilement sur Internet :
Gabedit : http://sites.google.com/site/allouchear/Home/gabedit/download
GAMESS : http://www.msg.ameslab.gov/gamess/download.html
La distribution de GAMESS est un peu particulière, vous aurez plusieurs étapes à effectuer pour avoir le fichier exécutable final. Préférez la version 32bits, qui marche dans tout les cas de figure.
Dans le dossier que vous récupérerez de GAMESS, se trouve un fichier PDF, qui vous guidera dans la marche à suivre pour l'installation de logiciels (HPC Pack 2008 R2 MS-MPI ainsi que Microsoft visual C++ 2010) nécessaires au fonctionnement de GAMESS (fichier WinGAMESS-setup-guide-32.pdf).
La suite de ce TP présentera les fonctions basiques apportées par Gabedit pour la construction de structure et la création des fichiers d'entrée GAMESS, avec la description des principaux mots-clés.
Question
Prise en main de Gabedit :
Ouvrez le logiciel Gabedit pour arriver sur l'interface principale.
La construction des différentes structures se fait par le biais de l'onglet Geometry>Draw
Vous arrivez ainsi sur cette nouvelle interface :
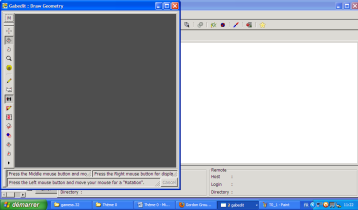
Sur le coté, vous disposez d'une barre d'outil vous permettant de créer votre molécule, soit en insérant des atomes, soit en insérant des fragments déjà pré-enregistrés dans le logiciel (bouton insert a fragment). Une nouvelle interface apparaît où vous aurez le choix entre de nombreux fragments. Enfin, la dernière option de la barre correspond à une mesure des distances, angles et angles dièdres de la molécule. C'est à partir de là que vous pouvez modifier la valeur des grandeurs mesurables. Vous pouvez aussi lire une structure d'un fichier de sortie GAMESS en faisant clic droit>Read>Gamess
Lorsque votre molécule est construite, fermez cette interface (la structure est automatiquement sauvegardée), puis cliquez, dans l'interface principale, sur l'icône GAMESS en dessous de l'onglet File, pour ouvrir l'interface dédiée à la création du fichier d'entrée (input) GAMESS.
L'interface est comme ceci :
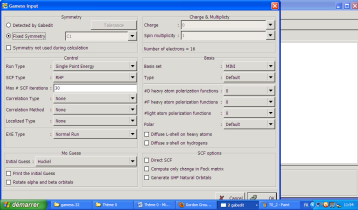
La plupart du temps, vous utiliserez l'option « Symmetry not used during calculation », surtout lorsque vous ne connaissez pas le groupe de symétrie de la molécule que vous étudiez. Sinon, en cochant « detected by Gabedit », le groupe le plus pertinent vous est tout de suite proposé. L'option Run type contient tout les types de calculs que vous allez faire (optimisation, calcul d'énergie, fréquence, recherche d'états de transition...). Enfin à droite, vous pouvez choisir la base que vous désirez pour le calcul. Il existe beaucoup d'autres fonctions pour la préparation d'un input GAMESS que vous pouvez modifier à loisir mais qui sortent du contexte de ce mini-didacticiel.
Une fois que vous avez choisi vos options, l'input GAMESS apparaît sur l'interface principale lorsque vous appuyez sur OK en bas de la fenêtre. Voici un exemple d'un input GAMESS obtenu :
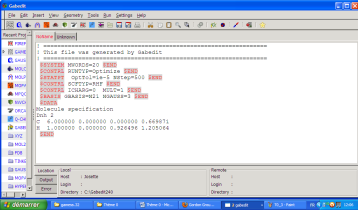
Différents mots-clés sont apparus en fonction de vos demandes. Nous allons les détailler rapidement :
- $SYSTEM correspond aux paramètres systèmes nécessaires pour le calcul, comme par exemple l'allocation de mémoire. Ici MWORDS correspond à la mémoire demandée pour le calcul. Dans tous les cas, l'option mis par défaut est suffisante pour tous les calculs abordés dans les TP suivants.
- $CONTRL correspond à la section de contrôle du calcul, notamment les options liées au niveau de calcul. RUNTYP correspond au type de calcul, SCFTYP au type de méthode numérique demandée pour la résolution du cycle SCF. Enfin ICHARG définit la charge portée par la molécule et MULT sa multiplicité.
- $STATPT apparaît uniquement lorsque vous demandez un RUNTYP de type optimisation de géométrie (« optimize ») ou recherche d'états de transition (« Sadpoint »). On précisera le nombre de cycles SCF ainsi que le critère de convergence.
- $BASIS contrôle les paramètres du jeu de fonctions de bases.
Après la sauvegarde du fichier dans le répertoire où se trouve le fichier exécutable GAMESS, vous devez ouvrir l'invite de commande de Windows (menu Démarrer >Exécuter > cmd). Allez dans le répertoire en question pour pouvoir lancer le calcul avec la commande :
rungms.bat fichier_entrée.inp 11-32 nb_procs nb_noeud fichier_sortie.log.
11-32 correspond à la version de l'exécutable GAMESS que vous possédez. La plupart du temps, lancer le calcul sur 1 seul processeur et 0 nœud (Cela est généralement le cas lorsque vous calculez sur un PC et non sur une grille de calcul) est largement suffisant pour les calculs que vous serez amené à effectuer. Voici un exemple pour un calcul sur l'éthylène :
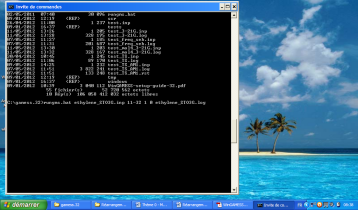
Lorsque le calcul est terminé, le fichier de sortie est directement ouvert par Gabedit si le fichier d'entrée est toujours ouvert par celui-ci. Vous arrivez alors sur cette interface en cliquant sur le fichier .log :
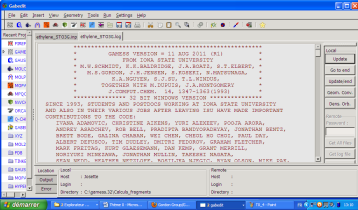
Sur votre droite, se trouvent des options sur ce fichier. Si le calcul est une optimisation de géométrie, l'option Geom.Conv. vous donnera les différents points obtenus par le calcul ainsi que le minimum global trouvé.
L'option Dens.Orb. est à sélectionner après un calcul d'énergie. Cette option ouvre une nouvelle interface où peuvent être visualisées les orbitales moléculaires, les fréquences de vibration et les mouvements des atomes qui leurs sont associées, etc...
L'interface est comme ceci :
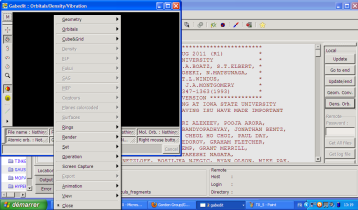
Le menu apparaît avec un clic droit. Vous devrez tout d'abord recharger en mémoire dans cette interface le fichier .log désiré pour la visualisation des orbitales ou des modes de vibration. Exemple avec le cas des orbitales :
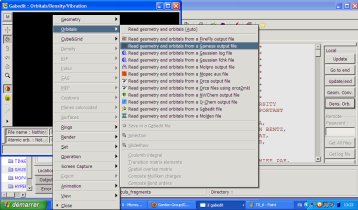
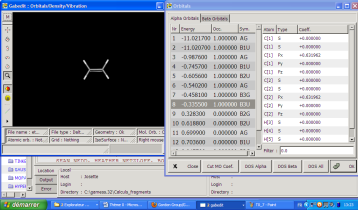
Apparaît alors l'écran de sélection des orbitales, avec leur énergie, leur symétrie et leur occupation. En cliquant sur une de ces orbitales, vous pouvez la faire apparaître sur l'écran. Considérons l'orbitale 8 de l'éthylène :
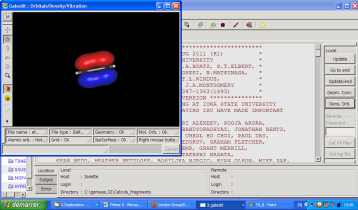
On peut refaire apparaître le menu de sélection des orbitales avec clic droit > orbitals > selection.
Solution détaillée
Avec ce petit didacticiel, vous êtes maintenant prêt pour réaliser les exercices proposés. Ils sont au nombre de 6 et permettant de découvrir par la pratique la chimie quantique et les diverses possibilités qu'elle offre (orbitales atomiques/moléculaires, la géométrie des molécules, la réactivité de composés...).